
Stuck on an issue?
Lightrun Answers was designed to reduce the constant googling that comes with debugging 3rd party libraries. It collects links to all the places you might be looking at while hunting down a tough bug.
And, if you’re still stuck at the end, we’re happy to hop on a call to see how we can help out.
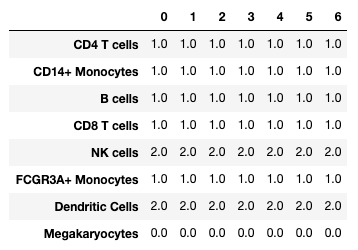
sc.tl.marker_gene_overlap() give the same values to each cluster
See original GitHub issueDear There,
I recently re-installed the single cell environment. Then, the example code of sc.tl.marker_gene_overlap() on scanpy tutorial does not work. It give the same values to each cluster. Thanks for your help!
Lianyun
import scanpy as sc
adata = sc.datasets.pbmc68k_reduced()
sc.pp.pca(adata, svd_solver='arpack')
sc.pp.neighbors(adata)
sc.tl.louvain(adata)
sc.tl.rank_genes_groups(adata, groupby='louvain')
marker_genes = {'CD4 T cells': {'IL7R'},
'CD14+ Monocytes': {'CD14', 'LYZ'},
'B cells': {'MS4A1'},
'CD8 T cells': {'CD8A'},
'NK cells': {'GNLY', 'NKG7'},
'FCGR3A+ Monocytes': {'FCGR3A', 'MS4A7'},
'Dendritic Cells': {'FCER1A', 'CST3'},
'Megakaryocytes': {'PPBP'}}
marker_matches = sc.tl.marker_gene_overlap(adata, marker_genes)
0 1 2 3 4 5 6
CD4 T cells 1.0 1.0 1.0 1.0 1.0 1.0 1.0
CD14+ Monocytes 1.0 1.0 1.0 1.0 1.0 1.0 1.0
B cells 1.0 1.0 1.0 1.0 1.0 1.0 1.0
CD8 T cells 1.0 1.0 1.0 1.0 1.0 1.0 1.0
NK cells 2.0 2.0 2.0 2.0 2.0 2.0 2.0
FCGR3A+ Monocytes 1.0 1.0 1.0 1.0 1.0 1.0 1.0
Dendritic Cells 2.0 2.0 2.0 2.0 2.0 2.0 2.0
Megakaryocytes 0.0 0.0 0.0 0.0 0.0 0.0 0.0
Versions
anndata 0.7.4 scanpy 1.6.0 sinfo 0.3.1
PIL 7.2.0 anndata 0.7.4 backcall 0.2.0 cffi 1.14.1 cycler 0.10.0 cython_runtime NA dateutil 2.8.1 decorator 4.4.2 get_version 2.1 h5py 2.10.0 igraph 0.8.2 ipykernel 5.3.4 ipython_genutils 0.2.0 jedi 0.15.2 joblib 0.16.0 kiwisolver 1.2.0 legacy_api_wrap 1.2 llvmlite 0.34.0 louvain 0.7.0 matplotlib 3.3.1 mpl_toolkits NA natsort 7.0.1 numba 0.51.2 numexpr 2.7.1 numpy 1.19.1 packaging 20.4 pandas 1.1.1 parso 0.5.2 pexpect 4.8.0 pickleshare 0.7.5 pkg_resources NA prompt_toolkit 3.0.7 ptyprocess 0.6.0 pycparser 2.20 pygments 2.6.1 pyparsing 2.4.7 pytz 2020.1 scanpy 1.6.0 scipy 1.5.2 setuptools_scm NA sinfo 0.3.1 six 1.15.0 sklearn 0.23.2 statsmodels 0.12.0 storemagic NA tables 3.6.1 texttable 1.6.3 tornado 6.0.4 traitlets 4.3.3 umap 0.4.6 wcwidth 0.2.5 zmq 19.0.2
IPython 7.18.1 jupyter_client 6.1.7 jupyter_core 4.6.3 notebook 6.1.3
Python 3.8.5 | packaged by conda-forge | (default, Aug 29 2020, 01:22:49) [GCC 7.5.0] Linux-3.10.0-1062.9.1.el7.x86_64-x86_64-with-glibc2.10 128 logical CPU cores, x86_64
Session information updated at 2020-09-09 15:47
Issue Analytics
- State:

- Created 3 years ago
- Comments:7 (2 by maintainers)
 Top Results From Across the Web
Top Results From Across the Web
sc.tl.marker_gene_overlap() give the same values to each ...
I recently re-installed the single cell environment. Then, the example code of sc.tl.marker_gene_overlap() on scanpy tutorial does not work. It ...
Read more >scanpy.tl.marker_gene_overlap - Read the Docs
The method returns a pandas dataframe which can be used to annotate clusters based on marker gene overlaps. This function was written by...
Read more >scanpy_05_dge
Once we have done clustering, let's compute a ranking for the highly differential genes in each cluster. Differential expression is performed with the ......
Read more >Cell-Wise Marker Gene Overlap - R-Project.org
Description. Calculates the per-cell overlap of previously calculated marker genes. ... which cells have the same markers, regardless of clustering?
Read more >Comparing samples within a merged dataset - Help - Scanpy
Hi, I have a dataset composed of 2 samples, one is control and the ... out how to use sc.tl.rank_genes_groups to compare the...
Read more > Top Related Medium Post
Top Related Medium Post
No results found
 Top Related StackOverflow Question
Top Related StackOverflow Question
No results found
 Troubleshoot Live Code
Troubleshoot Live Code
Lightrun enables developers to add logs, metrics and snapshots to live code - no restarts or redeploys required.
Start Free Top Related Reddit Thread
Top Related Reddit Thread
No results found
 Top Related Hackernoon Post
Top Related Hackernoon Post
No results found
 Top Related Tweet
Top Related Tweet
No results found
 Top Related Dev.to Post
Top Related Dev.to Post
No results found
 Top Related Hashnode Post
Top Related Hashnode Post
No results found
Thanks @jpreall and @suzhanhao for jumping in with solutions here. I have finally found some time to look at this. The recent change is that
sc.tl.rank_genes_groupsnow saves DE results not just for the top 100 genes, but for all genes. So the default fortop_n_markers, which was to use all genes that have results stored automatically shifted from 100 genes to all genes. And if you look at overlaps between all genes and your gene list, you will of course get 1. The only change that is needed here would be to set the defaulttop_n_markers = 100. That should be a 5 character change to the function 😉.Addressed by #1464