
Lightrun Answers was designed to reduce the constant googling that comes with debugging 3rd party libraries. It collects links to all the places you might be looking at while hunting down a tough bug.
And, if you’re still stuck at the end, we’re happy to hop on a call to see how we can help out.
scikit-misc cannot be initiated when sc.pp.highly_variable_genes(adata, n_top_genes=5000, flavor='seurat_v3')
See original GitHub issue- I have checked that this issue has not already been reported.
- I have confirmed this bug exists on the latest version of scanpy.
- (optional) I have confirmed this bug exists on the master branch of scanpy.
Hello Scanpy, When I’m running sc.pp.highly_variable_genes(adata, n_top_genes=5000, flavor=‘seurat_v3’), it asks me to install scikit-misc, which is already installed. Please see the picture below. Could you please help me to solve this issue? Thanks! Best, YJ Note: Please read this guide detailing how to provide the necessary information for us to reproduce your bug.
Minimal code sample (that we can copy&paste without having any data)
# Your code here
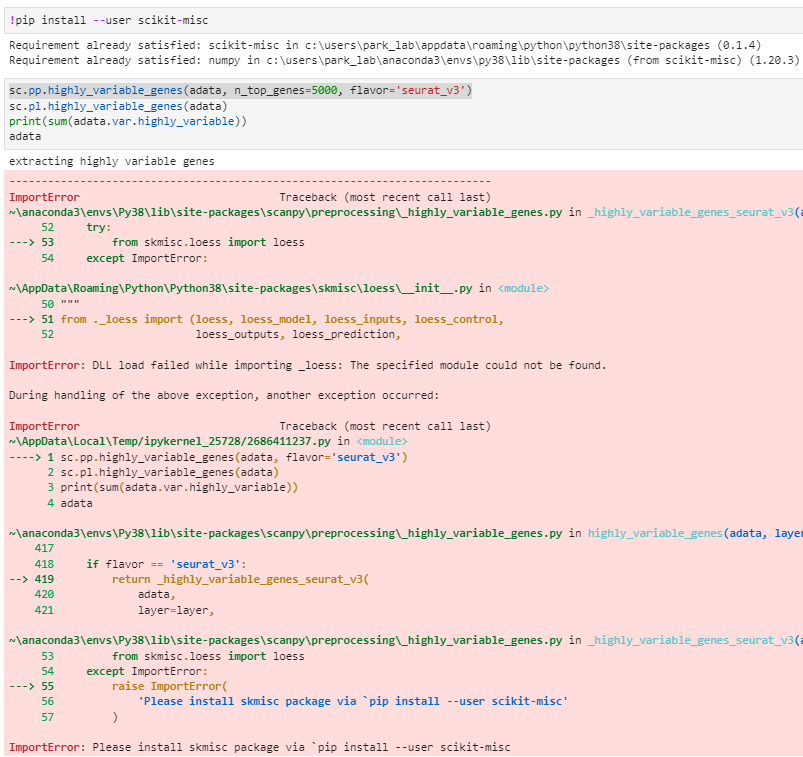
[Paste the error output produced by the above code here]
Versions
[Paste the output of scanpy.logging.print_versions() leaving a blank line after the details tag]
Issue Analytics
- State:

- Created 2 years ago
- Comments:14 (3 by maintainers)
 Top Related StackOverflow Question
Top Related StackOverflow Question Troubleshoot Live Code
Troubleshoot Live Code Top Related Reddit Thread
Top Related Reddit Thread Top Related Hackernoon Post
Top Related Hackernoon Post Top Related Tweet
Top Related Tweet Top Related Dev.to Post
Top Related Dev.to Post Top Related Hashnode Post
Top Related Hashnode Post
I found a workaround that does not require downloading the
.whlfile fornumpy=1.19.5. By default, MKL is included when you install numpy with conda. It’s good to do this in a new environment.Now I can run
sc.pp.highly_variable_genes()with no problem.Hello @davidhbrann, Thanks for the suggestion. I found that
numpy‑1.19.5+mkl‑cp36‑cp36m‑win_amd64.whlonly supports python 3.6, but the support of python 3.6 has been dropped by Scanpy. I didn’t findnumpy-1.20.3+mkl-cp38-cp38-win_amd64.whlby google yet, but I believe that your solution will work. It seems thatnumpy‑1.21.5+mkl‑cp38‑cp38‑win_amd64.whlis the only hope, but Scanpy requires numpy<=1.20. I think it’s the compatibility issue. Hope Scanpy can solve this compatibility issue in the future. Thanks! Best, YJ