
Lightrun Answers was designed to reduce the constant googling that comes with debugging 3rd party libraries. It collects links to all the places you might be looking at while hunting down a tough bug.
And, if you’re still stuck at the end, we’re happy to hop on a call to see how we can help out.
Seurat uses log-transformed and scaled data for analysis, Scanpy uses raw, which method is better?
See original GitHub issue… Hello Scanpy, In @LuckyMD 's amazing paper (https://www.embopress.org/doi/full/10.15252/msb.20188746), Table 1 shows that using raw data to calculate the maker genes of clusters is the appropriate way. But the raw data was not regressed out with mitochondrial genes, gene counts, cell cycle scores…So there will be so many mito genes ranked on the top of the marker gene list. What shall we do with these mito genes, because usually they represent the dead cell-released RNA contaminations?
In Seurat, they did every downstream analysis and plotting by using the log-transformed and scaled data (see below, the scaled dots in Seurat violin plot). Scanpy draws all plots by setting use_raw=True. I’m wondering which method is better?
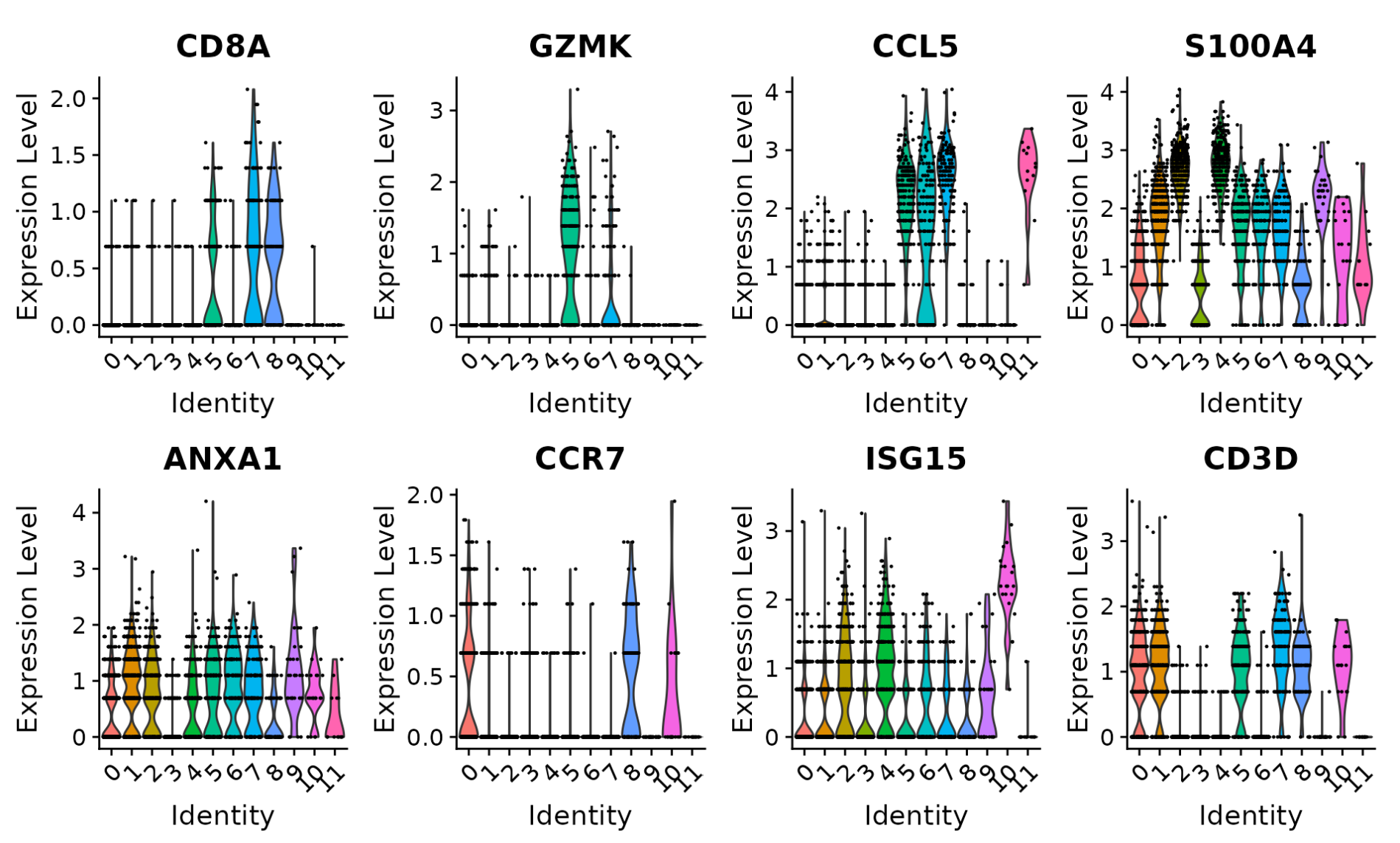
BTW, logFC will become negative and disappear for the marker genes of clusters when we set use_raw=False in sc.tl.rank_genes_groups(adata, 'leiden', method='wilcoxon'. Please check this https://github.com/theislab/scanpy/issues/2057.
Thanks! Best, YJ
Issue Analytics
- State:

- Created 2 years ago
- Comments:5 (3 by maintainers)
 Top Related StackOverflow Question
Top Related StackOverflow Question Troubleshoot Live Code
Troubleshoot Live Code Top Related Reddit Thread
Top Related Reddit Thread Top Related Hackernoon Post
Top Related Hackernoon Post Top Related Tweet
Top Related Tweet Top Related Dev.to Post
Top Related Dev.to Post Top Related Hashnode Post
Top Related Hashnode Post
Because adata was regressed, gene expression will become negative, cannot be loged.
If you have to regress out covariates, maybe you could do it after log transformation? I’m not 100% sure about this approach either though.